
Suche nach umweltbedingten Ursachen schreitet voran
Es wird allgemein angenommen, dass Umweltexpositionen für die große Mehrheit der sporadischen Parkinson-Krankheit (PD) verantwortlich sind, entweder allein oder durch Interaktionen mit genetischen Faktoren. Die Suche nach umweltbedingten Ursachen von PD wurde jedoch durch das mangelnde Verständnis der prodromalen Phase der PD-Entwicklung und der Schwierigkeiten bei der Expositionsabschätzung während dieses längeren Zeitraums behindert. Andererseits bietet das Vorhandensein dieser Prodromalperiode zusammen mit einem zunehmend besseren Verständnis der PD-Prodromalsymptome eine aufregende Gelegenheit, Umweltfaktoren zu identifizieren, die die PD-Pathogenese initiieren und / oder deren Verlauf modifizieren. Für die Präventionsbemühungen ist diese Prodromalphase von großem Interesse. Targeting-Faktoren, die über die Nase oder den Darm in den Körper gelangen, sind seit der Entdeckung von α- Synuclein-Aggregaten im enterischen und olfaktorischen Nervensystem noch wichtiger geworden. In diesem Artikel spekulieren wir über neuartige Forschungshypothesen und -ansätze, die uns dabei helfen können, die Rolle der Umwelt in der PD-Ätiologie insbesondere in ihrer erweiterten und komplexen prodromalen Phase besser zu definieren.
EINFÜHRUNG
Die Ursachen für sporadisch auftretende Parkinson-Krankheit (PD), die zu einem späten Zeitpunkt einsetzt, sind nach wie vor unklar, und die PD ist wahrscheinlich das kumulative Ergebnis zahlreicher genetischer und Umweltbelastungen und ihrer Wechselwirkungen im Zusammenhang mit der Gehirnalterung. Die Erforschung der Umweltauslöser und -modifikatoren für die PD-Entwicklung ist aus einer Reihe von Gründen äußerst wichtig. Erstens dauert die Entwicklung einer sporadischen PD zu einem späteren Zeitpunkt Jahrzehnte, und zum Zeitpunkt der Diagnose sind neurodegenerative Veränderungen zu weit fortgeschritten, um sie zu verlangsamen, zu stoppen oder umzukehren. Unser Kampf gegen die PD hängt daher entscheidend von der Früherkennung und Intervention von Krankheiten ab, die wiederum auf einem guten Verständnis der Krankheitsvernichtung und auf veränderbaren Risikofaktoren beruhen. Zweitens, trotz der jüngsten großen Erfolge bei der Entdeckung der genetischen Basis der sporadischen PD mit spätem Beginn, können genetische Befunde nur einen kleinen Teil der Fälle erklären und können nicht ohne weiteres auf die Prävention von Krankheiten ausgedehnt werden. Auf der anderen Seite können in den Jahrzehnten der PD im Prodromalstadium viele Umweltfaktoren zu verschiedenen Zeitpunkten ins Spiel kommen, die eine PD-Pathogenese auslösen und deren Progression verändern können [ 1 ]. Es ist vernünftig anzunehmen, dass in einer Mehrheit, wenn nicht in allen, erst spät auftretenden PD-Fällen Umweltbeiträge vorliegen, die das Risiko und das Alter des PD-Beginns bestimmen oder verändern. Leider sind wir weit davon entfernt, diese Faktoren zu identifizieren, ihre Rollen zu definieren und ihre Beiträge zu quantifizieren. Drittens sind Umweltfaktoren im Gegensatz zu genetischen Faktoren potentiell modifizierbar und werden daher tiefgreifende Auswirkungen auf die Prävention und Behandlung von PD haben. Angesichts des rapiden Wachstums der alternden Bevölkerung auf der ganzen Welt stellt sich die PD schließlich als die am schnellsten wachsende neurologische Erkrankung sowohl hinsichtlich der Prävalenz als auch des Todes heraus [ 2, 3 ]. Neben dieser potenziellen Krise im Bereich der öffentlichen Gesundheit legen die jüngsten, wenn auch vorläufigen und inkonsistenten Beweise nahe, dass es in den letzten Jahrzehnten einen zunehmenden Trend bei der Inzidenz von PD gibt [ 4 ], was möglicherweise eine Rolle für Umweltfaktoren darstellt. Zusammengenommen besteht ein dringender Handlungsbedarf der Finanzierungsstellen und der PD-Forscher, um Umweltbeiträge zur Entwicklung der PD zu ermitteln.
WAS WISSEN WIR JETZT ÜBER UMWELTFAKTOREN UND PD?
In den letzten zwei Jahrzehnten haben Wissenschaftler mehr als ein Dutzend Umweltfaktoren identifiziert, die mit dem Risiko der Entwicklung einer PD zusammenhängen, und für die Mehrheit sind die Ergebnisse zwischen den Studien einigermaßen konsistent [ 1, 5 ]. Beispiele sind inverse Assoziationen mit dem Rauchen [ 6, 7 ], Kaffeetrinken [ 8, 9 ], kräftiger Bewegung [ 10, 11 ], Ibuprofen-Verwendung [ 12, 13 ] und Plasma-Urat [ 14, 15 ] sowie positiven Assoziationen bei einer Pestizidexposition insgesamt [ 16, 17 ], Verwendung spezifischer Pestizide [ 18, 19 ] und traumatischer Hirnverletzung [ 20, 21 ]. Für die meisten dieser Assoziationen wurden plausible biologische Hypothesen vorgeschlagen. Die kausale Inferenz dieser epidemiologischen Befunde war jedoch sehr schwierig. Abgesehen von begrenzten und häufig inkonsistenten experimentellen Daten ist die umgekehrte Kausalität für die meisten dieser epidemiologischen Beobachtungen eine mögliche Erklärung dafür – dass die Entwicklung der PD vor der klinischen Diagnose den Lebensstil und das Verhalten ändert und nicht umgekehrt. Mögliche Ausnahmen sind die Verwendung bestimmter Pestizide. Zum Beispiel werden epidemiologische Befunde zu Rotenon und Paraquat [ 18, 19 ] durch starke experimentelle Nachweise gestützt, so dass diese Chemikalien zur Erzeugung von Nagetiermodellen für die PD-therapeutische Forschung verwendet werden [ 22 ]. Selbst für Pestizide gibt es viele wichtige Fragen, die nicht beantwortet werden. Trotz seiner Bedeutung und einer vernünftigen Anhäufung von Literatur steckt unser Verständnis von Umweltbeiträgen für die PD noch in den Kinderschuhen.
WAS SIND DIE WICHTIGSTEN HERAUSFORDERUNGEN, UM UMWELTFREUNDLICHKEITEN VON PD ZU SUCHEN?
Die Tatsache, dass sich die sporadische PD zu einem späteren Zeitpunkt erst nach Jahrzehnten entwickelt, und das mangelnde Verständnis für diese verlängerte prodromale Phase stellt eine große Herausforderung dar, um den Beitrag der Umwelt zur PD zu verstehen. In diesem Krankheitsentwicklungsparadigma müssen möglicherweise ursächliche Expositionen auftreten, die einen PD-pathologischen Prozess auslösen, und müssen Jahrzehnte vor der klinischen Diagnose der Erkrankung dokumentiert werden, was in epidemiologischen Studien oft nicht durchführbar ist. Sobald die Neurodegeneration initiiert ist, können viele umweltbedingte und genetische Faktoren dazu beitragen, das Fortschreiten der PD während der Jahrzehnte der Entwicklung prodromaler Erkrankungen zu verändern. Infolgedessen sind auch die robustesten epidemiologischen Befunde, einschließlich der Langzeitkohorten, alternativen Erklärungen unterworfen. Zum Beispiel haben Raucher in allen Arten von epidemiologischen Studien ein robustes und wesentlich niedrigeres Risiko für PD als Nicht-Raucher [ 23 ]. Während eine kausale Interpretation, dass Zigarettenrauchen das PD-Risiko reduziert, ansprechend ist, sind alternative Hypothesen wie umgekehrte Kausalität und Verwirrung durch die Persönlichkeit und andere unbekannte Risikofaktoren ebenfalls möglich [ 24 ]. Ähnliche Analogien können leicht auf die meisten, wenn nicht alle, der oben genannten mutmaßlichen „schützenden“ modifizierbaren Risikofaktoren erweitert werden.
Eine weitere große Herausforderung ist das Fehlen eines guten Verständnisses von „Umwelt“, das ein breites Spektrum von Expositionen umfasst, mit denen Menschen in Kontakt kommen, von Chemikalien (z. B. Pestiziden) über physikalische Wirkstoffe, Mikroben und Viren bis hin zu Klima, Lebensstil und sozioökonomischen Faktoren Bedingungen und Interaktionen mit der Host-Umgebung. Im Gegensatz zum menschlichen Genom haben wir keinen „Bauplan“ unserer Umwelt; Zusätzlich zur Komplexität ändern sich die Umweltexpositionen im Laufe der Zeit, und ihre gesundheitlichen Folgen sind wahrscheinlich komplex, kumulativ, interaktiv und dynamisch. Während dies bei fast allen chronischen Krankheiten in der Umweltforschung häufige Herausforderungen sind, sind sie insbesondere bei neurodegenerativen Erkrankungen wie PD eine Herausforderung. Neuronen sind langlebig und daher sind die gleichen alternden Neuronen während der gesamten Lebensdauer potenziellen Umwelteinflüssen ausgesetzt. Des Weiteren wird die PD nicht mehr als Erkrankung des Gehirns betrachtet, sondern umfasst mehrere Systeme und Organe. Bei der Untersuchung von Umweltbeiträgen zur PD müssen Umweltbelastungen durch mehrere Eintrittspfade und durch mehrere Anfälligkeitsfenster berücksichtigt werden, die noch genauer definiert werden müssen. All dies, zusammen mit einem jahrzehntelangen Beginn der Erkrankung und einer prodromalen Entwicklungszeit, macht eine zuverlässige und valide Expositionsabschätzung in epidemiologischen Studien, die die relevantesten ätiologischen Perioden erfasst, sehr schwer zu erreichen.
JETZT IST EINE GROSSE GELEGENHEIT, UM NACH VORN ZU BEWEGEN!
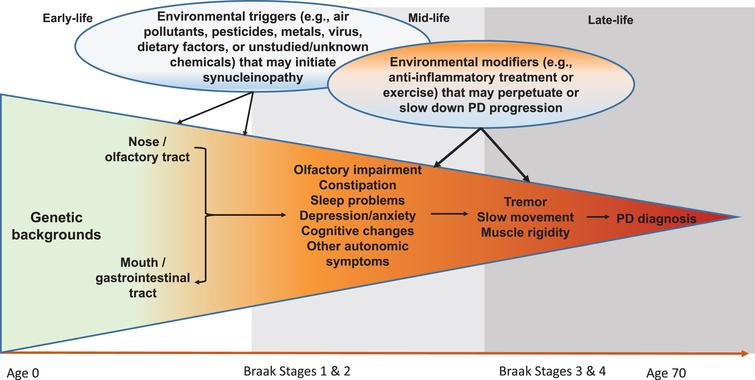
Die Braak-Hypothese [ 25 ] ist zwar immer noch etwas umstritten, stellt jedoch einen einzigartigen hypothetischen Rahmen für die PD-Entwicklung dar, der es uns ermöglicht, Schritte der prodromalen PD-Entwicklung und Umweltbeiträge besser zu konzeptualisieren. Gemäß dieser Hypothese entwickelt sich die PD-Lewy-Pathologie in sechs aufeinanderfolgenden Stadien, zuerst im Riechkolben oder in den Darmnerven (Stadium 1), Jahre, wenn nicht Jahrzehnte, bevor sie sich auf die Substantia nigra ausbreitet, in der der Tod dopaminerger Neurone auftritt (Stadium 3). Zur Unterstützung dieser Hypothese haben kürzlich durchgeführte klinische und epidemiologische Studien ein breites Spektrum von nichtmotorischen Symptomen bei PD-Patienten und einige Symptome wie Riechstörungen [ 26, 27 ], REM-Schlafstörung (RBD) [ 28, 29 ] eindeutig dokumentiert. Verstopfung [ 30, 31 ] kann Jahre, wenn nicht Jahrzehnte vor der klinischen Diagnose der PD entwickelt haben. Während es immer noch erhebliche Herausforderungen gibt, die prodromale PD adäquat zu definieren, können wir durch Verwendung dieser Symptome als nichtinvasive intermediäre Phänotypen neue Einblicke in diese „Blackbox“ der prodromalen Entwicklung von PD bringen, indem Faktoren identifiziert werden, die eine PD-Pathogenese auslösen diese intermediären Phänotypen oder modifizieren den Fortschritt zur klinischen PD ( 1 ). Dieses Rahmenwerk kann das Verständnis der PD-Entwicklung und der Beiträge von Umweltfaktoren grundlegend verbessern.
Fig.1
Systematischer und lebenslanger Ansatz zur Untersuchung von Umweltauslösern und -modifikatoren für die PD-Entwicklung. Sporadische PD mit spätem Beginn braucht Jahrzehnte, um sich zu entwickeln. In der frühen oder mittleren Lebenszeit können einige Umweltgifte (z. B. Pestizide, Luftschadstoffe, Viren) über die Nase oder den Mund in den Körper gelangen, was bei anfälligen Individuen über Mechanismen wie Entzündungen oder Mikrobiomendysbiose pathologische Synukleinopathie hervorrufen kann. Im Laufe der Zeit kann sich die Pathologie zu den zentralen Riechstrukturen und / oder zum unteren Hirnstamm entwickeln und Symptome wie Riechstörungen und Schlafstörungen hervorrufen. Ein Teil der Betroffenen kann im Laufe der Jahre weitere motorische Defizite entwickeln, die schließlich zu einer PD-Diagnose führen können. Während dieses langwierigen Prozesses können zu verschiedenen Zeitfenstern viele Umweltfaktoren eine Rolle spielen, um das Fortschreiten der Krankheit fortzusetzen oder zu verlangsamen. Modifiziert nach Chen H (2018). J Parkinsons Dis. 8 , 1–12.
NEUE UMWELTPERSPEKTIVEN
Die Braak-Hypothese impliziert den Riechweg und den Verdauungstrakt als mögliche Ursprünge der PD-Entwicklung [ 25 ]. Es ist wichtig, dass dies die zwei anatomischen Stellen sind, an denen der menschliche Körper direkt mit der Umgebung interagiert, an denen häufig Entzündungen auftreten und die Wege zum Gehirn gut etabliert sind. Es ist möglich, dass Umweltgifte, wie Luftschadstoffe, Pestizide, Verunreinigungen aus der Nahrung oder Viren, über die Nase und / oder den Mund in den menschlichen Körper gelangen und die PD-Pathogenese am Riechkolben und / oder den Darmdarmnerven initiieren können. Im Laufe der Zeit kann sich die Pathogenese über den Riech- und / oder Vagusnerv im Gehirn ausbreiten und letztendlich zu Todesfällen von dopaminergen Neuronen in der Substantia nigra führen [ 32 ]. Alle diese Beobachtungen führen Untersuchungen der Umweltexpositionen durch, die über den Riech- oder Verdauungstrakt eintreten und für die PD-Ätiologie relevant und attraktiv sind. Im Folgenden werden einige gemeinsame und relevante Exposures von Interesse erörtert.
Luftverschmutzer
Der olfaktorische Weg stellt einen Eintrittspunkt für Luftschadstoffe dar, der die Blut-Hirn-Schranke (BBB) umgeht, und erweist sich als Weg für die Übertragung pathogener α- Synucleins in das Gehirn [ 33 ]. Es ist daher plausibel, zu vermuten, dass Luftschadstoffe zur Pathogenese der PD beitragen können, indem sie eine α- Synuclein-Neuropathologie im Riechsystem initiieren, die später im Rahmen der Gehirnalterung zur PD übergehen kann [ 33 ]. Epidemiologische Nachweise zu Luftschadstoffen und zur PD sind provokativ, aber begrenzt und etwas inkonsistent [ 34–37 ]. Interessanterweise wurden α- Synuclein-Aggregate im Riechkolben von Kleinkindern, Kindern, Jugendlichen und jungen Erwachsenen beobachtet, die in Mexiko-Stadt, wo die Luftverschmutzung hoch ist, vorzeitig starben [ 38 ]. In den USA, wo die Luftschadstoffgehalte wesentlich niedriger sind, wurden in jüngster Zeit Luftschadstoffe mit einer Beeinträchtigung des Riechstoffs bei älteren Erwachsenen in Verbindung gebracht [ 39, 40 ]. Dies wird begleitet von einer wachsenden Literatur über mögliche schädliche Auswirkungen von Luftschadstoffen auf die kognitive Funktion und Demenz [ 41 ] sowie auf Neuroinflammation als einen potenziellen Weg zwischen Luftschadstoffen und neurodegenerativen Ergebnissen [ 42 ]. Wir erwarten zukünftige Studien, die nicht nur Luftschadstoffe, Geruchsstörungen und die Entwicklung der PD verbinden, sondern auch die zugrunde liegenden biologischen Mechanismen ermitteln.
Das Darmmikrobiom
Das Darmmikrobiom wurde schnell als ein Hauptfaktor für die Physiologie des Menschen erkannt, da es das Immunsystem beeinflusst, und es ist für die Aufnahme von Nährstoffen, Medikamenten und Umweltgiften verantwortlich. Es gibt immer mehr Beweise dafür, dass das Mikrobiom verschiedene Aspekte neurologischer Funktionen, Hirnaktivitäten und Verhaltensweisen sowohl in Tiermodellen als auch beim Menschen beeinflussen kann [ 43 ]. Die Braak-Hypothese stellt das Mikrobiom außerdem in die vorderste Reihe der PD-Erforschung [ 43, 44 ], da es Darm-Darmnerven als Initiationsstelle der PD-Pathologie impliziert [ 25 ]. Um dies zu unterstützen, ist Verstopfung eines der häufigsten prodromalen Symptome von PD [ 45 ], das sich 1-2 Jahrzehnte vor der PD-Diagnose entwickelt haben kann [ 30, 31, 46 ]. Neuere Studien haben die Darmmikrobiome von PD-Patienten mit denen von Kontrollen verglichen [ 47–53 ]. Obwohl die Ergebnisse nicht vollständig übereinstimmten, identifizierten die Studien im Vergleich zu Kontrollen im Allgemeinen ein proinflammatorisches Mikrobiom bei PD-Patienten. Interessanterweise berichtete eine kürzlich veröffentlichte Studie über Ähnlichkeiten bei der Mikrobiomdysbiose zwischen PD und idiopathischen RBD-Patienten, was darauf schließen lässt, dass sich das Mikrobiom bei der prodromalen PD ändern kann [ 53 ]. Da das Darmmikrobiom durch viele PD-relevante Umweltfaktoren (z. B. Rauchen, Ernährungsfaktoren und Pestizide) beeinflusst werden kann, sind Untersuchungen zur Untersuchung potenzieller Umwelteinflüsse auf das Darmmikrobiom im Rahmen der PD-Entwicklung erforderlich. Ähnliche Analogien ließen sich auch leicht mit den nasalen oder oralen Mikrobiomen machen, die in der PD-Forschung bisher weitgehend ignoriert wurden [ 53, 54 ].
Neue Einblicke in die Entwicklung von Pestiziden und PD
Pestizide gehören zu den wenigen gut dokumentierten schädlichen Umwelteinflüssen für die PD [ 18, 19 ]. Wann, wo und wie bestimmte Pestizide zur Entwicklung von PD beitragen, ist jedoch weitgehend unbekannt. Pestizide gelangen durch Einatmen, Essen / Trinken oder Hautkontakt in den Körper [ 55 ]; Daher können die Geruchs- oder Verdauungs-Eintrittspunkte leicht auf Pestizidexpositionen ausgedehnt werden. Wenn Pestizide über die Nase oder den Verdauungstrakt in den Körper gelangen, können sie eine Synukleinopathie in der Riechstruktur oder im Darm auslösen, die sich später im Laufe der Zeit über den Riechnerv oder die Darm-zu-Gehirn-Achse ausbreiten kann [ 56 ]. Die Erforschung von Pestiziden in Bezug auf Verstopfung und Beeinträchtigung des Geruchssinns im Zusammenhang mit dem Alter kann Informationen liefern, die für die Entwicklung der PD von entscheidender Bedeutung sind. Obwohl Riech- und Magen-Darm-Symptome in PD-Tiermodellen auf Pestizidbasis dokumentiert wurden [ 57, 58 ], ist die empirische Evidenz des Menschen spärlich und indirekt. Eine Studie in North Carolina fand heraus, dass Landarbeiter höhere Riechschwellen hatten als Nichtlandarbeiter [ 59 ], aber die Studie konnte diese Beobachtung nicht direkt auf den Einsatz von Pestiziden zurückführen. Interessanterweise deuten jüngste Beweise darauf hin, dass einige Pestizide das Darmmikrobiom verändern. In Mausmodellen störte das Organophosphat-Insektizid Diazinon die Gemeinschaftsstruktur, das funktionelle Metagenom und das Stoffwechselprofil des Darmmikrobioms [ 60, 61 ] durch Modulation des Quorum Sensing, einem Schlüsselmechanismus, der die Bakterienpopulation, die Zusammensetzung und vor allem ihre funktionellen Gene reguliert . Wir erwarten in Zukunft detaillierte Untersuchungen, um unser Verständnis der Rolle spezifischer Pestizide bei der PD-Entwicklung zu verbessern, indem mögliche Eintrittswege, Auswirkungen auf verschiedene Stadien der PD-Entwicklung und mögliche biologische oder pathologische Mechanismen beschrieben werden.
Andere relevante Umweltexpositionen
Obwohl die Braak-Hypothese nicht im Mittelpunkt dieses Artikels steht, bietet sie auch klare Gründe, um systematisch mehrere andere Umweltexpositionen zu untersuchen, die im Zusammenhang mit der PD-Entwicklung nicht gut untersucht wurden, wie organische Lösungsmittel [ 62 ], gekochtes Fleisch mit hoher Temperatur und heterocyclische Amine [ 63 ], respiratorische oder gastrointestinale Infektionen und Entzündungen [ 64–66 ] sowie den Einsatz von Antibiotika und antiviralen Therapien [ 67 ].
WEITERE WICHTIGE ÜBERLEGUNGEN BEI DER FESTLEGUNG VON UMWELTBEWERTUNGEN FÜR DIE PD
Gen-Umwelt-Interaktionen und Epigenetik
Viele biologische Prozesse haben sich durch mehrere Mechanismen entwickelt, die sowohl genetische als auch ökologische Beiträge beinhalten, und beide müssen zusammenkommen, bevor ein biologisches System ausfällt. Die Identifizierung genetischer Faktoren, die die Assoziation spezifischer Umweltstressoren mit PD beeinflussen, wird unser Verständnis ätiologischer Mechanismen verbessern. Der Erfolg beim Verständnis der PD-Gen-Umwelt-Wechselwirkungen war bisher weitgehend auf Wechselwirkungen genetischer Faktoren mit spezifischen Pestiziden beschränkt. Studien weisen zum Beispiel darauf hin, dass die potenziellen nachteiligen Auswirkungen von Paraquat auf die PD durch genetische Varianten, die mit der Dopamin-Transporterfunktion [ 68 ] oder dem Ausfall der DNA-Basen-Exzisionsreparatur [ 69 ] zusammenhängen, möglicherweise erheblich verstärkt werden, möglicherweise indem das System nicht in der Lage ist, dies auszugleichen die kombinierten synergistischen Beleidigungen. Während diese Einzelstudien entscheidende Anhaltspunkte für das Verständnis spezifischer Gen-Umwelt-Wechselwirkungen und -Pfade liefern, erwarten wir von zunehmend verfügbaren und statistisch leistungsfähigeren gepoolten Analysen, [ 70, 71 ] potentielle Gen-Umwelt-Wechselwirkungen zu identifizieren, zu überprüfen oder zu widerlegen [ 72 ] PD-Ätiologie.
Die epigenetische Forschung ist ein weiterer aufregender Ansatz zum Verständnis der PD-Pathogenese, indem Mechanismen untersucht werden, die die Genfunktion durch DNA-Methylierung und Histon-Modifikation regulieren, ohne die DNA-Sequenz selbst zu verändern. Die meisten PD-epigenetischen Studien zielten bisher auf Methylierung in Kandidatengenen mit Blut- oder Speichelproben ab [ 73 ], und die Interpretation der Studienergebnisse ist durch die Tatsache eingeschränkt, dass die epigenetischen Vorschriften oft gewebespezifisch sind. Aktuelle epigenomweite Assoziations- und Netzwerkanalysen zeigten jedoch, dass der PD-Status mit Änderungen der DNA-Methylierung in Blut und Speichel zusammenhängt, die das Immunsystem beeinträchtigten. 74, 75 ]. Diese Beobachtung konvergiert gut mit den zunehmenden genomischen [ 76, 77 ], epidemiologischen [ 66 ] und experimentellen [ 78 ] Beweisen für die Bedeutung von Immunität und Entzündung bei der PD-Entwicklung. Epigenetische Ansätze sowie andere „omics“ – Werkzeuge (Proteinomics, Transcriptomics, Lipidomics, Metabolomics), die auf menschlichen Gewebeproben und induzierten humanen Stammzellen basieren, können ebenfalls für den „meet in the middle“ – Ansatz verwendet werden Integration solcher Technologien in die epidemiologische Forschung [ 79 ]. Wir erwarten, dass solche synergistischen Ansätze mehr Einblick in die biologische Plausibilität ermöglichen und zur Ermittlung der Kausalität epidemiologischer Befunde beitragen.
Ein lebenslanger und exponentieller Ansatz für die ätiologische Forschung von PD
Die meisten epidemiologischen Studien zu PD konzentrierten sich auf eine Momentaufnahme einer einzigen Umweltexposition, die sich häufig im mittleren bis späten Erwachsenenalter befand, wobei die multidimensionale Komplexität der Krankheit, die eine Lebenslaufperspektive erfordern könnte, weitgehend ignoriert wurde. Einige experimentelle Studien haben gezeigt, dass eine vorgeburtliche oder frühzeitige Exposition gegenüber bestimmten Pestiziden (z. B. Dieldrin [ 80 ], Paraquat oder Maneb [ 81 ], Atrazin [ 82 ]) später im Leben zu einer dopaminergen Neuronendegeneration führen kann. Andere berichteten, dass solche Expositionen im frühen Lebensalter die Tiere anfälliger für eine zweite toxische Umweltbelastung im späteren Leben machten [ 81, 83 ]. Diese Ergebnisse legen nahe, dass Expositionen im frühen Lebensalter zur Entwicklung der PD beitragen können, indem sie das Stadium der Anfälligkeit für PD in der frühen Lebensphase vorsieht, und weisen darauf hin, wie wichtig es ist, wiederholte Expositionen während anfälliger Perioden zu verhindern. Obwohl ein lebenslanger Ansatz für spät auftretende neurodegenerative Erkrankungen wie die PD methodisch enorm ist, erwarten wir, dass zukünftige Studien diese Möglichkeiten sorgfältig prüfen.
Die jüngste genetische Forschung hat begonnen, einen polygenen Risiko-Score zu generieren, der alle bekannten Risiko-Allele umfasst, und seine Verwendung wurde gefördert, um die allgemeine genetische Anfälligkeit eines Individuums für die PD zu definieren [ 84 ] oder die Vorhersage des Krankheitsverlaufs [ 85 ]. Im Vergleich dazu ist die Entwicklung eines „zusammengesetzten Umweltindex“ viel schwieriger, und die große Mehrheit der epidemiologischen Studien konzentrierte sich auf einen einzigen Risikofaktor (z. B. Rauchen). Trotzdem haben epidemiologische Studien erste Anstrengungen unternommen, um mehrere Umweltbelastungen gleichzeitig zu berücksichtigen. Zum Beispiel haben Lee et al. berichteten über mögliche Gelenkeffekte aufgrund einer traumatischen Hirnverletzung und Paraquat-Exposition beim PD-Risiko [ 86 ] und Kim et al. gleichzeitig berücksichtigt wurden Rauchen, Koffeinkonsum, körperliche Aktivität und verschiedene andere Faktoren [ 87 ]. Diese Ergebnisse sind provokativ und deuten auf stärkere Assoziationen bei der Berücksichtigung zusammengesetzter Risikopositionen und deren möglichen Synergismus hin. Darüber hinaus werden Biomarker für langfristige und lebenslange Umweltexpositionen entwickelt, z. B. Blut-Methylierungsprofile für langfristige Organophosphat-Expositionen [ 88 ]. Darüber hinaus wurden einige wichtige wissenschaftliche und technologische Fortschritte erzielt, die die externe Beurteilung des Exposoms beeinflussen [ 91, 92 ], einschließlich geografischer Informationssysteme, Fernerkundung, globaler Ortungssysteme und Geolokalisierungstechnologien, tragbare und persönliche Erfassung, einschließlich Sensoren auf Smartphone-Basis und selbstausgelesene Fragebogenbewertungen, die auf internetbasierten Plattformen basieren. Alle diese methodischen und technologischen Verbesserungen bei der Expositionsbewertung müssen jedoch mit neuen Datenanalyse- und Interpretationsmethoden einhergehen und erfordern auch die Entwicklung von Protokollen für den ethischen Austausch sensibler Daten.
Wenn es darum geht, mehrere Expositionen über einen längeren Zeitraum hinweg zu berücksichtigen, sind die Begriffe „Exposom“ und „Neuroexposom“ besonders ansprechend. Chris Wild schlug das Konzept des „Exposoms“ vor mehr als einem Jahrzehnt erstmals als Maß für die Gesamtheit der menschlichen Umweltexpositionen während des gesamten Lebens vor [ 89 ]. Heffernan und Hare [ 90 ] haben dieses Konzept kürzlich für neurologische Erkrankungen angepasst. Sie schlugen vor, die Stärken der „Omics“ -Technologien (z. B. Genomics und Metabolomics), traditionelle epidemiologische Erhebungen und detaillierte klinische Bewertungen über die gesamte Lebensdauer hinweg zu kultivieren und dabei die einzigartigen Merkmale des Gehirns zu berücksichtigen (z. B. Permeabilität von BBB und Langlebigkeit von Neuronen).
Dieses Exposomenkonzept ist zwar schwierig zu implementieren in der realen Welt und bietet zusammen mit der Braak-Hypothese einen theoretischen Rahmen für Wissenschaftler, um zukünftige Studien zu entwerfen, um die Umweltursachen von PD zu entschlüsseln und frühzeitige Interventionen zu entwickeln, um die Entwicklung der charakteristischen motorischen Dysfunktion aufzuhalten PD.
Quelle: Journal Of Parkinson’s Desease
Gastredakteure: Patrik Brundin, J. William Langston und Bastiaan R. Bloem
Autoren: Chen, Honglei a ; * | Ritz, Beate geb.
Zugehörigkeiten: [ a ] Abteilung für Epidemiologie und Biostatistik, Hochschule für Humanmedizin, Michigan State University, East Lansing, MI, USA | [ b ] Abteilung für Epidemiologie und Umweltgesundheit, Fielding School of Public Health, Universität von Kalifornien, Los Angeles, Los Angeles, Kalifornien, USA
Journal: Journal of Parkinson , vol. 8, nein. s1, S. S9-S17, 2018 Akzeptiert am 15. November 2018 | Veröffentlicht am: 18. Dezember 2018
Übersetzt mit Hilfe von „Google Übersetzer“
INTERESSENKONFLIKT
Der Autor hat keinen Interessenkonflikt zu melden.
BESTÄTIGUNGEN
Dr. Chen wird durch einen Start-up-Fonds der Michigan State University (GE100455), der Parkinson-Stiftung (Grant-Nr. PF-IMP-1825) und des Büros des stellvertretenden Verteidigungsministers für Gesundheitswesen durch die Parkinson-Forschung unterstützt Programm (Auszeichnung Nr. W81XWH-17-1-0536). Meinungen, Interpretationen, Schlussfolgerungen und Empfehlungen sind die des Autors und werden nicht unbedingt vom Verteidigungsministerium gebilligt. Dr. Ritz wurde von den NIEHS-Zuschüssen ES10544, P01ES016732, U54ES12078, R01ES013717, R21ES022391, R21ES024356 und dem NINDS-Zuschuss: P50NS038367 und durch Pilotfinanzierung von SCEHSC # 5P30 ES07048 und American Parkinson Disease Association unterstützt. Wir möchten uns auch bei Herrn Frank Purdy für das Korrekturlesen des Manuskripts bedanken.
REFERENCES
[1]
Chen H (2018) The changing landscape of Parkinson epidemiologic research. J Parkinsons Dis 8, 1–12.
[2]
GBD 2016 Parkinson’s Disease Collaborators (2018) Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17, 939–953.
[3]
Darweesh SKL , Raphael KG , Brundin P , Matthews H , Wyse RK , Chen H , Bloem BR (2018) Parkinson Matters. J Parkinsons Dis 8, 495–498.
[4]
Savica R , Grossardt BR , Bower JH , Ahlskog JE , Rocca WA (2017) Time trends in the incidence of Parkinson’s disease: A 30-year study. JAMA Neurol 32, 227–234.
[5]
Ascherio A , Schwarzschild MA (2016) The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol 15, 1257–1272.
[6]
Chen H , Huang X , Guo X , Mailman RB , Park Y , Kamel F , Umbach DM , Xu Q , Hollenbeck A , Schatzkin A , Blair A (2010) Smoking duration, intensity, and risk of Parkinson disease. Neurology 74, 878–884.
[7]
Ritz B , Lee PC , Lassen CF , Arah OA (2014) Parkinson disease and smoking revisited: Ease of quitting is an early sign of the disease. Neurology 83, 1396–1402.
[8]
Ascherio A , Weisskopf MG , O’Reilly EJ , McCullough ML , Calle EE , Rodriguez C , Thun MJ (2004) Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: The modifying effects of estrogen. Am J Epidemiol 160, 977–984.
[9]
Liu R , Guo X , Park Y , Huang X , Sinha R , Freedman ND , Hollenbeck AR , Blair A , Chen H (2012) Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am J Epidemiol 175, 1200–1207.
[10]
Chen H , Zhang SM , Schwarzschild MA , Hernan MA , Ascherio A (2005) Physical activity and the risk of Parkinson disease. Neurology 64, 664–669.
[11]
Xu Q , Park Y , Huang X , Hollenbeck A , Blair A , Schatzkin A , Chen H (2010) Physical activities and future risk of Parkinson disease. Neurology 75, 341–348.
[12]
Chen H , Jacobs E , Schwarzschild MA , McCullough ML , Calle EE , Thun MJ , Ascherio A (2005) Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 58, 963–967.
[13]
Gao X , Chen H , Schwarzschild MA , Ascherio A (2011) Use of ibuprofen and risk of Parkinson disease. Neurology 76, 863–869.
[14]
Chen H , Mosley TH , Alonso A , Huang X (2009) Plasma urate and Parkinson’s disease in the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol 169, 1064–1069.
[15]
Gao X , O’Reilly EJ , Schwarzschild MA , Ascherio A (2016) Prospective study of plasma urate and risk of Parkinson disease in men and women. Neurology 86, 520–526.
[16]
Ascherio A , Chen H , Weisskopf MG , O’Reilly E , McCullough ML , Calle EE , Schwarzschild MA , Thun MJ (2006) Pesticide exposure and risk for Parkinson’s disease. Ann Neurol 60, 197–203.
[17]
Liew Z , Wang A , Bronstein J , Ritz B (2014) Job exposure matrix (JEM)-derived estimates of lifetime occupational pesticide exposure and the risk of Parkinson’s disease. Arch Environ Occup Health 69, 241–251.
[18]
Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade AR , Comyns K , Richards MB , Meng C , Priestley B , Fernandez HH , Cambi F , Umbach DM , Blair A , Sandler DP , Langston JW (2011) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119, 866–872.
[19]
Costello S , Cockburn M , Bronstein J , Zhang X , Ritz B (2009) Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169, 919–926.
[20]
Fang F , Chen H , Feldman AL , Kamel F , Ye W , Wirdefeldt K (2012) Head injury and Parkinson’s disease: A population-based study. Mov Disord 27, 1632–1635.
[21]
Gao J , Liu R , Zhao E , Huang X , Nalls MA , Singleton AB , Chen H (2015) Head injury, potential interaction with genes, and risk for Parkinson’s disease. Parkinsonism Relat Disord 21, 292–296.
[22]
Le W , Sayana P , Jankovic J (2014) Animal models of Parkinson’s disease: A gateway to therapeutics? Neurotherapeutics 11, 92–110.
[23]
Breckenridge CB , Berry C , Chang ET , Sielken RL Jr , Mandel JS (2016) Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS One 11, e0151841.
[24]
Ritz B , Rhodes SL (2010) After half a century of research on smoking and PD, where do we go now? Neurology 74, 870–871.
[25]
Braak H , Bohl JR , Müller CM , Rüb U , de Vos RAI , Tredici KD (2006) Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord 21, 2042–2051.
[26]
Chen H , Shrestha S , Huang X , Jain S , Guo X , Tranah GJ , Garcia ME , Satterfield S , Phillips C , Harris TB , Health ABCS (2017) Olfaction and incident Parkinson disease in US white and black older adults. Neurology 89, 1441–1447.
[27]
Ross GW , Petrovitch H , Abbott RD , Tanner CM , Popper J , Masaki K , Launer L , White LR (2008) Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol 63, 167–173.
[28]
Postuma RB , Gagnon JF , Vendette M , Fantini ML , Massicotte-Marquez J , Montplaisir J (2009) Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology 72, 1296–1300.
[29]
Postuma RB , Iranzo A , Hogl B , Arnulf I , Ferini-Strambi L , Manni R , Miyamoto T , Oertel W , Dauvilliers Y , Ju YE , Puligheddu M , Sonka K , Pelletier A , Santamaria J , Frauscher B , Leu-Semenescu S , Zucconi M , Terzaghi M , Miyamoto M , Unger MM , Carlander B , Fantini ML , Montplaisir JY (2015) Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: A multicenter study. Ann Neurol 77, 830–839.
[30]
Abbott RD , Petrovitch H , White LR , Masaki KH , Tanner CM , Curb JD , Grandinetti A , Blanchette PL , Popper JS , Ross GW (2001) Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 57, 456–462.
[31]
Gao X , Chen H , Schwarzschild MA , Ascherio A (2011) A prospective study of bowel movement frequency and risk of Parkinson’s disease. Am J Epidemiol 174, 546–551.
[32]
Reichmann H (2011) View point: Etiology in Parkinson’s disease. Dual hit or spreading intoxication. J Neurol Sci 310, 9–11.
[33]
Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VM , Brundin P (2016) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213, 1759–1778.
[34]
Ritz B , Lee PC , Hansen J , Lassen CF , Ketzel M , Sorensen M , Raaschou-Nielsen O (2016) Traffic-related air pollution and Parkinson’s disease in Denmark: A case-control study. Environ Health Perspect 124, 351–356.
[35]
Liu R , Young MT , Chen JC , Kaufman JD , Chen H (2016) Ambient air pollution exposures and risk of Parkinson disease. Environ Health Perspect 124, 1759–1765.
[36]
Lee PC , Liu LL , Sun Y , Chen YA , Liu CC , Li CY , Yu HL , Ritz B (2016) Traffic-related air pollution increased the risk of Parkinson’s disease in Taiwan: A nationwide study. Environ Int 96, 75–81.
[37]
Palacios N , Fitzgerald KC , Hart JE , Weisskopf M , Schwarzschild MA , Ascherio A , Laden F (2017) Air pollution and risk of Parkinson’s disease in a large prospective study of men. Environ Health Perspect 125, 087011.
[38]
Calderon-Garciduenas L , Gonzalez-Maciel A , Reynoso-Robles R , Kulesza RJ , Mukherjee PS , Torres-Jardon R , Ronkko T , Doty RL (2018) Alzheimer’s disease and alpha-synuclein pathology in the olfactory bulbs of infants, children, teens and adults </=40 years in Metropolitan Mexico City. APOE4 carriers at higher risk of suicide accelerate their olfactory bulb pathology. Environ Res 166, 348–362.
[39]
Ajmani GS , Suh HH , Wroblewski KE , Kern DW , Schumm LP , McClintock MK , Yanosky JD , Pinto JM (2016) Fine particulate matter exposure and olfactory dysfunction among urban-dwelling older US adults. Environ Res 151, 797–803.
[40]
Adams DR , Ajmani GS , Pun VC , Wroblewski KE , Kern DW , Schumm LP , McClintock MK , Suh HH , Pinto JM (2016) Nitrogen dioxide pollution exposure is associated with olfactory dysfunction in older U.S. adults. Int Forum Allergy Rhinol 6, 1245–1252.
[41]
Power MC , Adar SD , Yanosky JD , Weuve J (2016) Exposure to air pollution as a potential contributor to cognitive function, cognitive decline, brain imaging, and dementia: A systematic review of epidemiologic research. Neurotoxicology 56, 235–253.
[42]
Jayaraj RL , Rodriguez EA , Wang Y , Block ML (2017) Outdoor ambient air pollution and neurodegenerative diseases: The neuroinflammation hypothesis. Curr Environ Health Rep 4, 166–179.
[43]
Tremlett H , Bauer KC , Appel-Cresswell S , Finlay BB , Waubant E (2017) The gut microbiome in human neurological disease: A review. Ann Neurol 81, 369–382.
[44]
Parashar A , Udayabanu M (2017) Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat Disord 38, 1–7.
[45]
Chen H , Zhao EJ , Zhang W , Lu Y , Liu R , Huang X , Ciesielski-Jones AJ , Justice MA , Cousins DS , Peddada S (2015) Meta-analyses on prevalence of selected Parkinson’s nonmotor symptoms before and after diagnosis. Transl Neurodegener 4, 1.
[46]
Savica R , Carlin JM , Grossardt BR , Bower JH , Ahlskog JE , Maraganore DM , Bharucha AE , Rocca WA (2009) Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology 73, 1752–1758.
[47]
Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM (2015) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30, 1351–1360.
[48]
Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P (2015) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30, 350–358.
[49]
Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Burmann J , Fassbender K , Schwiertz A , Schafer KH (2016) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32, 66–72.
[50]
Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M (2015) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10, e0142164.
[51]
Petrov VA , Saltykova IV , Zhukova IA , Alifirova VM , Zhukova NG , Dorofeeva YB , Tyakht AV , Kovarsky BA , Alekseev DG , Kostryukova ES , Mironova YS , Izhboldina OP , Nikitina MA , Perevozchikova TV , Fait EA , Babenko VV , Vakhitova MT , Govorun VM , Sazonov AE (2017) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162, 734–737.
[52]
Hopfner F , Kunstner A , Muller SH , Kunzel S , Zeuner KE , Margraf NG , Deuschl G , Baines JF , Kuhlenbaumer G (2017) Gut microbiota in Parkinson disease in a northern German cohort. Brain Res 1667, 41–45.
[53]
Heintz-Buschart A , Pandey U , Wicke T , Sixel-Doring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P (2018) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33, 88–98.
[54]
Pereira PAB , Aho VTE , Paulin L , Pekkonen E , Auvinen P , Scheperjans F (2017) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat Disord 38, 61–67.
[55]
Mostafalou S , Abdollahi M (2017) Pesticides: An update of human exposure and toxicity. Arch Toxicol 91, 549–599.
[56]
Pan-Montojo F , Anichtchik O , Dening Y , Knels L , Pursche S , Jung R , Jackson S , Gille G , Spillantini MG , Reichmann H , Funk RH (2010) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5, e8762.
[57]
Sasajima H , Miyazono S , Noguchi T , Kashiwayanagi M (2015) Intranasal administration of rotenone in mice attenuated olfactory functions through the lesion of dopaminergic neurons in the olfactory bulb. Neurotoxicology 51, 106–115.
[58]
Johnson ME , Stringer A , Bobrovskaya L (2018) Rotenone induces gastrointestinal pathology and microbiota alterations in a rat model of Parkinson’s disease. Neurotoxicology 65, 174–185.
[59]
Quandt SA , Walker FO , Talton JW , Summers P , Chen H , McLeod DK , Arcury TA (2016) Olfactory function in Latino farmworkers: Subclinical neurological effects of pesticide exposure in a vulnerable population. J Occup Environ Med 58, 248–253.
[60]
Gao B , Bian X , Chi L , Tu P , Ru H , Lu K (2017) Editor’s Highlight: Organophosphate diazinon altered quorum sensing, cell motility, stress response, and carbohydrate metabolism of gut microbiome. Toxicol Sci 157, 354–364.
[61]
Gao B , Bian X , Mahbub R , Lu K (2017) Sex-specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ Health Perspect 125, 198–206.
[62]
Goldman SM , Quinlan PJ , Ross GW , Marras C , Meng C , Bhudhikanok GS , Comyns K , Korell M , Chade AR , Kasten M , Priestley B , Chou KL , Fernandez HH , Cambi F , Langston JW , Tanner CM (2012) Solvent exposures and Parkinson disease risk in twins. Ann Neurol 71, 776–784.
[63]
Agim ZS , Cannon JR (2018) Alterations in the nigrostriatal dopamine system after acute systemic PhIP exposure. Toxicol Lett 287, 31–41.
[64]
Huang HK , Wang JH , Lei WY , Chen CL , Chang CY , Liou LS (2018) Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Parkinsonism Relat Disord 47, 26–31.
[65]
Macerollo A , Lu MK , Huang HC , Chen HJ , Lin CC , Kao CH , Tsai CH , Chen JC (2017) Colonic diverticular disease: A new risk factor for Parkinson’s disease? Parkinsonism Relat Disord 42, 61–65.
[66]
Villumsen M , Aznar S , Pakkenberg B , Jess T , Brudek T (2018) Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut. 10.1136/gutjnl-2017-315666.
[67]
Racette BA , Gross A , Vouri SM , Camacho-Soto A , Willis AW , Searles Nielsen S (2018) Immunosuppressants and risk of Parkinson disease. Ann Clin Transl Neurol 5, 870–875.
[68]
Ritz BR , Manthripragada AD , Costello S , Lincoln SJ , Farrer MJ , Cockburn M , Bronstein J (2009) Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ Health Perspect 117, 964–969.
[69]
Sanders LH , Paul KC , Howlett EH , Lawal H , Boppana S , Bronstein JM , Ritz B , Greenamyre JT (2017) Editor’s Highlight: Base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol Sci 158, 188–198.
[70]
Chuang YH , Lee PC , Vlaar T , Mulot C , Loriot MA , Hansen J , Lill CM , Ritz B , Elbaz A (2017) Pooled analysis of the HLA-DRB1 by smoking interaction in Parkinson disease. Ann Neurol 82, 655–664.
[71]
Lee PC , Ahmed I , Loriot MA , Mulot C , Paul KC , Bronstein JM , Ritz B , Elbaz A (2018) Smoking and Parkinson disease: Evidence for gene-by-smoking interactions. Neurology 90, e583–e592.
[72]
Ahmed I , Lee PC , Lill CM , Searles Nielsen S , Artaud F , Gallagher LG , Loriot MA , Mulot C , Nacfer M , Liu T , Biernacka JM , Armasu S , Anderson K , Farin FM , Lassen CF , Hansen J , Olsen JH , Bertram L , Maraganore DM , Checkoway H , Ritz B , Elbaz A (2014) Lack of replication of the GRIN2A-by-coffee interaction in Parkinson disease. PLoS Genet 10, e1004788.
[73]
Miranda-Morales E , Meier K , Sandoval-Carrillo A , Salas-Pacheco J , Vazquez-Cardenas P , Arias-Carrion O (2017) Implications of DNA methylation in Parkinson’s disease. Front Mol Neurosci 10, 225.
[74]
Horvath S , Ritz BR (2015) Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 7, 1130–1142.
[75]
Chuang YH , Paul KC , Bronstein JM , Bordelon Y , Horvath S , Ritz B (2017) Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med 9, 76.
[76]
Witoelar A , Jansen IE , Wang Y , Desikan RS , Gibbs JR , Blauwendraat C , Thompson WK , Hernandez DG , Djurovic S , Schork AJ , Bettella F , Ellinghaus D , Franke A , Lie BA , McEvoy LK , Karlsen TH , Lesage S , Morris HR , Brice A , Wood NW , Heutink P , Hardy J , Singleton AB , Dale AM , Gasser T , Andreassen OA , Sharma M , International Parkinson’s Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) Investigators (2017) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol 74, 780–792.
[77]
Gagliano SA , Pouget JG , Hardy J , Knight J , Barnes MR , Ryten M , Weale ME (2016) Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol 3, 924–933.
[78]
Kannarkat GT , Boss JM , Tansey MG (2013) The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis 3, 493–514.
[79]
Vineis P , van Veldhoven K , Chadeau-Hyam M , Athersuch TJ (2013) Advancing the application of omics-based biomarkers in environmental epidemiology. Environ Mol Mutagen 54, 461–467.
[80]
Richardson JR , Caudle WM , Wang M , Dean ED , Pennell KD , Miller GW (2006) Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson’s disease. FASEB J 20, 1695–1697.
[81]
Cory-Slechta DA , Thiruchelvam M , Barlow BK , Richfield EK (2005) Developmental pesticide models of the Parkinson disease phenotype. Environ Health Perspect 113, 1263–1270.
[82]
Li Y , Sun Y , Yang J , Wu Y , Yu J , Li B (2014) Age-dependent dopaminergic dysfunction following fetal exposure to atrazine in SD rats. Environ Toxicol Pharmacol 37, 1275–1282.
[83]
Thiruchelvam M , Richfield EK , Goodman BM , Baggs RB , Cory-Slechta DA (2002) Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology 23, 621–633.
[84]
Nalls MA , Escott-Price V , Williams NM , Lubbe S , Keller MF , Morris HR , Singleton AB , International Parkinson’s Disease Genomics C (2015) Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 30, 850–854.
[85]
Paul KC , Schulz J , Bronstein JM , Lill CM , Ritz BR (2018) Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol 75, 360–366.
[86]
Lee PC , Bordelon Y , Bronstein J , Ritz B (2012) Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 79, 2061–2066.
[87]
Kim IY , O’Reilly EJ , Hughes KC , Gao X , Schwarzschild MA , Hannan MT , Betensky RA , Ascherio A (2018) Integration of risk factors for Parkinson disease in 2 large longitudinal cohorts. Neurology 90, e1646–e1653.
[88]
Paul K , Chuang Y , Cockburn M , Bronstein J , Horvath S , Ritz B (2018) Organophosphate pesticide exposure and differential genome-wide DNA methylation. Sci Total Environ 645, 1135–1143.
[89]
Wild CP (2005) Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev 14, 1847–1850.
[90]
Heffernan AL , Hare DJ (2018) Tracing Environmental exposure from neurodevelopment to neurodegeneration. Trends Neurosci 41, 496–501.
[91]
Turner MC , Nieuwenhuijsen M , Anderson K , Balshaw D , Cui Y , Dunton G , Hoppin JA , Koutrakis P , Jerrett M (2017) Assessing the exposome with external measures: Commentary on the state of the science and research recommendations. Annu Rev Public Health 38, 215–239.
[92]
Loh M , Sarigiannis D , Gotti A , Karakitsios S , Pronk A , Kuijpers E , Annesi-Maesano I , Baiz N , Madureira J , Oliveira Fernandes E , Jerrett M , Cherrie JW (2017) How sensors might help define the external exposome. Int J Environ Res Public Health 14.